The Gut: A Key to the Pathogenesis of Type 2-Diabetes?

Authored by Holst JJ
Mini Review
The gastrointestinal tract plays a predominant role in the regulation of the postprandial plasma glucose levels [1].
The first regulating factor is the gastric antral motility which, by
incompletely elucidated neuroendocrine mechanisms, regulates the rate of
emptying and hence exposure of chyme to the small intestine. Thus, it
is a main function of the stomach to receive and retain the incoming
meal, and eventually to expel the triturated, emulsified and partly
digested contents, the chyme, at a slow and surprisingly constant at a
rate of maximally 4Kcal per minute. The duration of gastric emptying
thereby becomes regulated by the energy content of the ingested meal [2].
The exposure rate of the small intestine is similarly limited and
relatively constant. If the regulatory mechanisms are interfered with
[for instance with surgical procedures like pyloroplasty or gastric
bypass) and the normal emptying rate is exceeded, a so-called "dumping
syndrome" may be elicited.
This syndrome which includes nausea, malaise, desire
to lie down, maybe fainting, is caused by the increased osmotic load
presented to the small intestine which shifts fluid from the circulation
to the intestinal lumen via the leaky epithelium in the proximal gut.
The more or less constant emptying rate also means that the secretion
and thereby the plasma concentration of some of the gut hormones, e.g.
the insulin-stimulating hormone, glucose-dependent insulin tropic
polypeptide [GIPJ which is dependent on the intestinal absorption rate
of nutrients, quickly rises to a certain elevated level which is then
maintained as long as there is emptying from the stomach, depending on
the total amount of stimulatory nutrients that were present in the meal
[and are being retained in the stomach) [2].
It is possible that hydrogen ions in the duodenum
[released from the stomach) play a role in the regulation of the
emptying rate, but nutrients in the duodenum may also inhibit the
emptying rate by neuroendocrine mechanisms. The hormone, GIP, has little
effect on gastric emptying, whereas the other incretin hormone,
glucagon-like peptide-1 [GLP-1), the secretion of which follows a
similar pattern, powerfully inhibits gastric emptying. There is no doubt
that the regulation of gastric emptying plays a major role for the
postprandial glucose responses, again as illustrated in conditions of
accelerated emptying rates, where particularly the early postprandial
glucose excursions may be dramatically increased [3].
The increased secretion of GIP and GLP-1 powerfully amplifies the
glucose-induced insulin secretion, and thereby also the ability of the
organism to limit and restore to normal the increased glucose
concentrations-indeed postprandial reactive hypoglycemia [sometimes
designated "late dumping") may ensue, and is relatively often observed
in individuals with accelerated gastric emptying [4].
The amplifying effect of the gut hormones on the
glucose- induced insulin secretion is designated the incretin effect,
and this is responsible for the proportional increase in insulin
secretion according to the ingested amount of glucose [and other meal
components). The effects is small after small meals [small amount of
glucose), but increases so that it may be responsible for up to 80% of
the elimination of glucose from the circulation after larger meals
[corresponding to 100g of glucose) [2,5].
By means of the regulation of gastric emptying and the incretin effect,
it is ensured that post prandial glucose excursions are normally
moderate and in fact independent of the ingested amount of calories
[glucose) [1].
It is likely that these mechanisms are of major
importance to prevent complications elicited by hyperglycemia. As
mentioned these powerful mechanisms may also lead to severe postprandial
reactive hypoglycemia [6].
The question then arises whether the same mechanisms play a role in the
development of type 2diabetes mellitus [T2DM)? The gastric emptying
rates in patients with T2DM show great variability and a consistent
abnormality do not seem to prevail (although abnormalities may very well
exist in selected groups).
The early response (0-60min) to ingestion of a mixed
meal of the two hormones, GIP and GLP-1, does not seem to be importantly
affected in patients with T2D, but GLP-1 secretion is more consistently
impaired in the later phase (from 60 minutes and onwards) [7]
and this may play a significant role. A similar impairment (and a
similar impairment of the incretin effect, see below) is seen in obesity
[8,9].
Probably it is of greater importance that the insulin tropic action of
both hormones is also impaired. During the progressive development of
glucose in tolerance, a gradual loss of the incretin effect, as well as
the insulin tropic action of the two hormones are observed [10]
and their effect is almost completely lost in full-blown diabetes as
far as physiological amounts of the hormones are concerned [11].
However, in contrast to the almost complete loss of
insulin tropic action of GIP, regardless of dose, suprahysiological
concentrations of GLP-1 may still have considerable effect [12].
Evidently, the loss of the incretin effect is of major importance for
the postprandial hyperglycemia of the patients with T2D and restoration
of the incretin effect with GLP-1-receptoragonists provides part of the
explanation for the antidiabetic effects of these agents. The
GLP-1receptoragonistsalso powerfully inhibits the gastric emptying rate,
at least acutely and this explains part of the effect on postprandial
glycaemia of the short-acting GLP- 1-receptor agonists (eventide and
lixisenatide).
Regarding the long-acting GLP-1-receptoragonists,
tachyphylaxis rapidly develops regarding their effects on gastric
emptying, while their incretin effect is preserved and when during meal
intake the plasma glucose concentration rises, they will exert their
incretin action, which at the cellular level in essence consists of
potentiating of the glucose-induced insulin secretion [13,14].
There is, however, an additional factor which plays a rather important
role: patients with T2DM typically have increased plasma concentrations
of glucagon, both in the fasting state and postprandial [7].
There is a general misconception that meal intake
should lead to inhibition of glucagon secretion; on the contrary, most
meals, in particular protein rich meals, will stimulate glucagon
secretion, but for patients with T2DM the increase is even larger
Because glucagon stimulates the hepatic glucose production, this rise
has considerable consequences for the postprandial glucose excursions,
as illustrated in experiments with glucagon receptor antagonists, which
effective lower both fasting and postprandial glucose levels [15].
But what is the explanation for the elevated postprandial levels of
glucagon in the patients with T2DM? Several mechanisms have been
proposed and they may all contribute. First of all, there are probably
signals from the GI-tract.
Several hormones might be of importance, including
GIP and GLP-2, which stimulate glucagon secretion and GLP-1, which
inhibits secretion. Their combined influence was investigated in a
systematic study where T2D patients received intravenous infusions of
each of these hormones (mimicking their postprandial plasma profiles)
superimposed on an intravenous glucose infusion mimicking the
concentration curves resulting from an OGTT [16].
Individually, it turned out, each of the hormones had the mentioned,
expected effects, but infused together the resulting glucagon secretion
profile was very similar to that observed after oral glucose alone.
Particularly GIP stimulated glucagon secretion. An increased activity of
GIP might therefore contribute to the postprandial hyperglucagonemia in
T2DM. This together with the loss of its insulin tropic effect in T2DM
might therefore suggest that GIP actually has diabetogenic effects [17].
However, it is also possible that the postprandial
hyperglucagonemia is derived not from the pancreas but from the GI-tract
where some of the endocrine cells, under certain circumstances, which
may include T2DM, may be able to produce glucagon. This is clearly seen
in individuals after total pancreatectomies, who exhibit a large
postprandial glucagon response after oral glucose [18]. It can actually be demonstrated that the response has important effects for hepatic glucose production.
Extra pancreatic glucagon may therefore contribute to
the diabetic hyperglucagonemia. Furthermore, there is strong evidence
from recent research that circulating amino acids play a predominant
role in the regulation of glucagon secretion, and in view of this, it
should always be suspected that altered amino acid levels underlie
clinical hyperglucagonemia. The reverse also seems to be true: that the
plasma level of glucagon plays a predominant role in the regulation of
the plasma levels of the amino acids; thus conditions with hyper- and
hypoglucagonaemiaare associated with low and high levels of plasma amino
acids, respectively [19].
Indeed, amino acid metabolism and the glucagon-producing alpha cells in
the pancreas seem to be coupled in a close, negative feed-back loop.
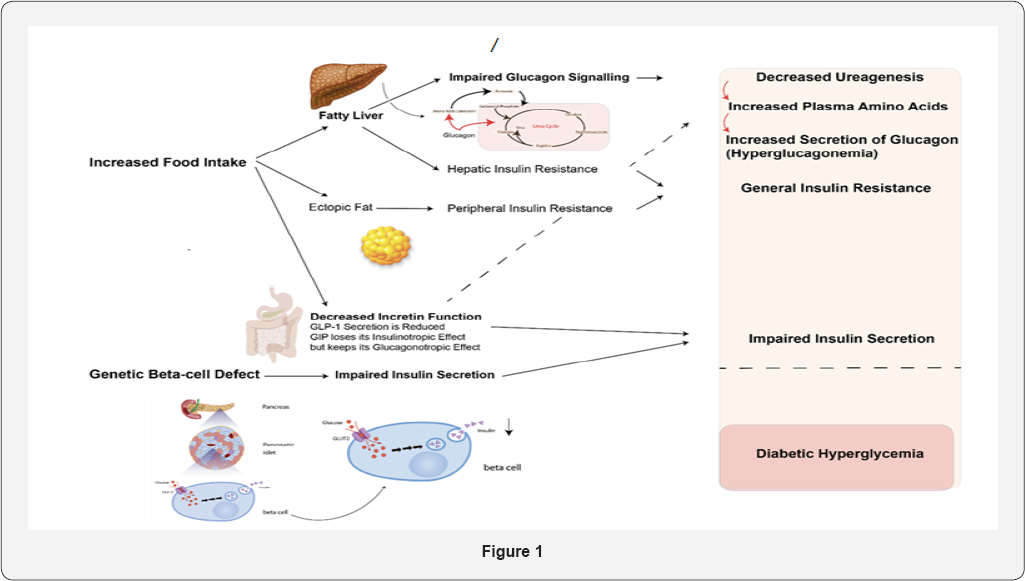
Most recently we examined patients with varying degrees of nonalcoholic
fatty lived disease and found a correlation between on one hand liver
function and amino acid levels and on the other hand the plasma glucagon
levels [20].
The hypothesis is that the steatosis impairs liver
function which also comprises an impairment of the effects of glucagon
on amino acid metabolism and urea genesis. As a consequence, plasma
levels of amino acids rise, which in turn results in hyperglucagonemia.
Because glucagon also influences glucose metabolism and hepatic glucose
production, this will also result in glucose intolerance and increased
postprandial glucose excursions. A lot of work still has to be done with
respect to unravelling these associations but the recent application of
glucagon receptor antagonists and the use of transgenic animals have
greatly facilitated these studies in both people and experimental
animals Figure 1 [21].

To Know More About Current Research in Diabetes & Obesity
Journal Please click on:
https://juniperpublishers.com/crdoj/index.php
https://juniperpublishers.com/crdoj/index.php


{kind=link}
Comments
Post a Comment